Images Collection

View this article in Search Friendly Plain Text
NOTE: This plain text article interpretation has been digitally created by OCR software to estimate the article text, to help both users and search engines find relevant article content. To read the actual article text, view or download the PDF above.
CARDIAC GLYCOSIDES AND CALCIUM
by
A. FARAH and P. N. WITT
Reprinted from
Proceedings of the First International Pharmacological Meeting ‘ Volume 3
PERGAMON PRESS
OXFORD ■ LONDON • NEW YORK • PARIS
1963
Reprinted from “Proceedings of the First International Pharmacological Meeting, Volume 3”
PERGAMON PRESS OXFORD • LONDON • NEW YORK • PARIS 1963
CARDIAC GLYCOSIDES AND CALCIUM
A. Farah and P. N. Witt
Department of Pharmacology, State University of New York, Upstate Medical Center, Syracuse, N.Y.
A number of theories purporting to explain digitalis action have been formulated. The indices of effects have been the changes in action potential, positive inotropic effect, irregularities, standstill contracture and oxygen consumption of the heart. It is not likely that all these stages are caused by one and the same mechanism. Thus a biochemical change produced by a certain concentration of a glycoside may not be related to all the pharmacological actions this glycoside can produce. In this discussion we considered all the cardiac glycosides to have basically the same mechanism differing only in their speed of action and elimination. This assumption was made since the data available in the literature were obtained with a variety of cardiac glycosides and in a variety of different systems and the qualitative effects were quite similar.
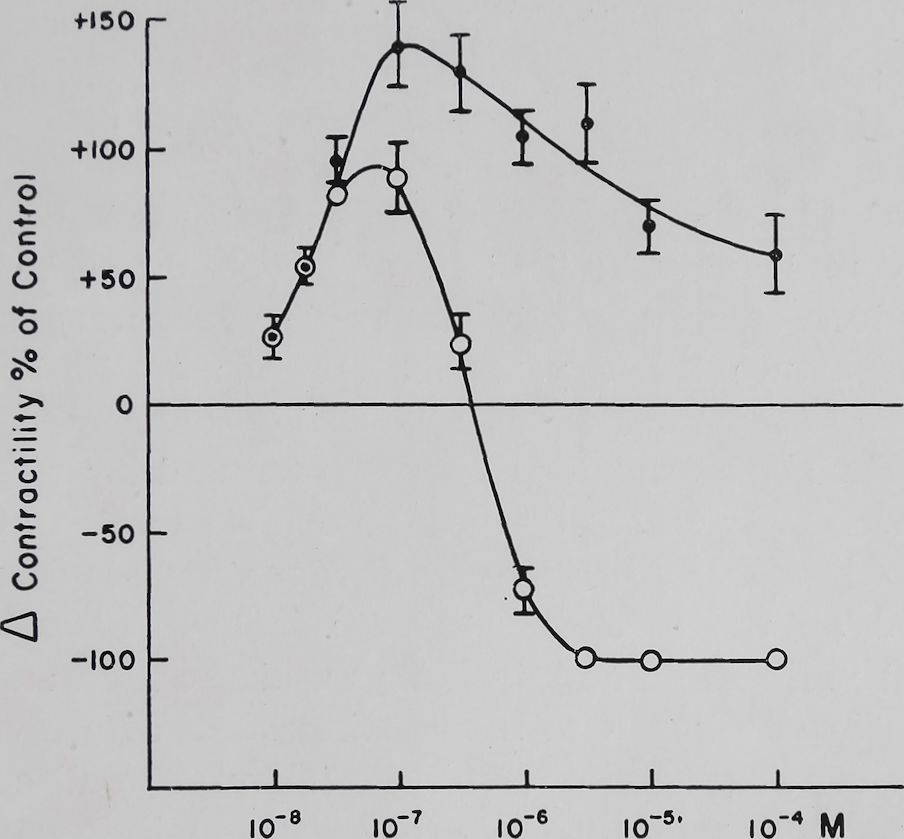
Stewart1 has published a dose-response curve for some cardiac glycosides on guinea pig ventricular strips. Similar results were obtained in rabbit auricular preparations with ouabain. Figure 1 shows that within a certain range of ouabain concentration the increase in contractility was proportional to the amount of this glycoside. Further increasing the concentration of ouabain reduced the positive inotropic effect. From his studies Stewart1 has concluded that this biphasic response was a resultant of inotropic and toxic processes with different rate constants. Increasing the concentration of glycoside increased the rate of appearance of toxic phenomena more than the rate of appearance of the positive inotropic effect. Thus high concentrations could produce toxic effects with minimal positive inotropic effects. The data presented in Fig. 1 show the hazards of correlating a biochemical response of a cardiac glycoside to its effect
[137]138
A. Farai-i and P. N. Witt
on contractility. The theory of Hajdu concerning digitalis action is a case in point and the essential aspects of this theory have been recently reviewed.2 In essence Hajdu states that the positive inotropic effect of cardiac glycosides is causally related to a reduction in intracellular monovalent ion concentration by reducing K+ influx. This hypothesis has been re-evaluated by Lee

Cone. Ouabain
Fig. 1. Effect of ouabain on the isolated left auricle of the rabbit stimulated at a constant rate of 4 per sec. The auricles were suspended in Tyrode solution containing in mM: NaCl 139, KC1 2-67, CaCl2 1-8, MgCl2 0*6, Na2HP04 0-4, NaHC03 12-0, and glucose 5-5. Temperature 37° C ± 0*1. Bath volume 50 ml. The auricles were suspended for 1 hr in the bath before addition of ouabain and then were followed for 90 min. Contractions were recorded isometri-cally by means of strain gauges on a Grass oscillograph utilizing d.c. preamplifiers. Maximum increase in contractility in per cent of control contractions +S.E. q—q Contractility 90 min after addition of ouabain. (Unpublished data, A. Farah and A. Caprio).
et al.3 and Tuttle et al.4 Both groups have shown that under tn vitro conditions the therapeutic effect of ouabain can be seen at a time when no het potassium loss above control can be demonstrated. Only with toxic doses of ouabain can one demon-
Cardiac glycosides and calcium
139
strate a potassium loss. The data of Tuttle et al.4 have also shown that so-called therapeutic doses of ouabain (2.5-5 X 10~8 M) may actually decrease the rate of loss of potassium thus confirming Boyer and Poindexter5 and decrease intracellular Na+ concentration thus confirming Clarke and Moscher6 who analyzed human cardiac tissue. Unpublished experiments of Tuttle and Witt have shown that therapeutic doses of ouabain which produced a good positive inotropic effect did not influence K42 influx or outflux while toxic doses inhibited this influx without affecting outflux. Infusions of a therapeutic dose of ouabain into intact anesthetized rabbits did not change intracellular K+ concentration but decreased the Na+ concentration. Amounts of ouabain which produced ventricular extrasystolies and runs of ventricular tachycardia did not change intracellular K+ or Na+ concentration. In those animals where ventricular fibrillation occurred a reduction of/intracellular K+ was observed (Table I). The data presented thus show that a positive inotropic effect produced by ouabain is probably not causally related to a reduction in intracellular K+ concentration nor to changes in K+ influx but these changes may be related to some of the severe toxic effects.
A relation between calcium ion and digitalis action has been suspected for some time. Thus Clark7 showed that digitoxin effects on the frog heart were not seen in the absence of calcium ion in the perfusion fluid and Ransom8 demonstrated that Strophanthin improved the contractility of the isolated frog heart made hypodynamic by reduction in the Ca++ concentration of the perfusion fluid. Based on experiments of Konschegg,9 Otto Loewi in 1918 published an important paper on this subject.10 He concluded that digitalis contracture of the frog heart was dependent on the presence of Ca++ in the bathing fluid and expressed the opinion that digitalis sensitized the heart to Ca++. In his review on digitalis action Lendle11 took issue with this interpretation and did not think that the evidence supported the Loewi hypothesis.
The findings that Ca++ decreased the lethal dose of digitalis 12—14 have been used as evidence for causally relating digitalis and Ca++ action. Wilbrandt15 in his masterful review on the relation of calcium to digitalis action cited experiments by Roh-rer which showed that short exposures of frog ventricular strips to digitalis sensitized the heart to calcium ion. Salter et al.1G
140
A. Farah and P. N. Witt
Table I
Effect of ouabain on K+ and Na+ in rabbit myocardium in situ
K+, Na+ and water content of rabbit hearts receiving saline, therapeutic, toxic or a lethal dose of ouabain. Rabbits 2-3 kg, pentobarbital anesthesia 30-45 mglkg. Ouabain given i.v. every 30 min, dose 0*05 or 0-025
mg/kg/30 min
| Tissue | No. of observations | Type of infusion | h2o
content (%) |
K+±S.E. (mEq/kg fiber wt.) | Na+ ± S.F. (mEq/kg fiber wt.) |
| Left atria | 10 | Saline
(control) |
84-3 | 71-2 ± 3-8 | 31-2 ± 3-6 |
| Left ventricles | 10 | 80-8 | 80-8 ± 0-8 | 22-1 ± 2-9 | |
| Left atria | 11 | Therapeutic dose of ouabain | 82-9 | 69-4 ± 2-5 | 24-5 ± 3-3 |
| Left ventricles | 11 | (0-1 mg/kg) | 79-5 | 80-3 ± 2-4 | 13-9 ± 2-5 |
| Left atria | 11 | Toxic dose of ouabain | 83-6 | 72-6 ± 2-6 | 27-2 ± 4-6 |
| Left ventricles | 10 | (0-2-0-25 mg/kg) irregularities | 81-1 | 80-9 ± 3-2 | 27-8 ± 4-9 |
| Left atria | 3 | Toxic dose of ouabain | 82-0 | 60-7 | 46-6 |
| Left ventricles | 3 | (0-2-0-25 mg/kg)
ventricular fibrillation |
81-1 | 59-9 | 58-0 |
have observed in frog hearts that ouabain action at relatively low Ca++ concentrations produced a percentile increase which was about the same at all Ca++ concentrations studied. Similar results have now been obtained in mammalian heart preparations. Table II shows that with Ca++ concentrations between 0*45 and 1-8 mM a therapeutic dose of ouabain (5 X 10—8M) produced an increase in contractility which was about the same at all these Ca++ levels. With concentrations of calcium from 1-8 to 7*2 mM the positive inotropic effect of ouabain was significantly decreased. If a higher concentration of ouabain (10-7 M) was used toxic manifestations were inhibited by low and appeared with
Cardiac glycosides and calcium
141
high calcium concentrations (Table II). All these findings suggested that digitalis in some way modified calcium action or calcium ion modified digitalis action.
Table II
Effect of Ca++ concentration on the inotropic effect of ouabain
Rabbit left auricles. Tyrode solution. Bath volume 50 ml. Temp. 37°C. Rate of stimulation = 3/sec. Control period with normal Tyrode = 45 min. Ouabain added 30 min after changing over to experimental solution. Rate = 3/sec. Each value is the average change ± S.E. obtained from
8-16 experiments
| Ca++ cone, in Tyrode solution (m M/l.) | Contractility during experimental period (% of control) | Effect of ouabain on contractility as % of experimental contractility | |||
| 5 X 10-8M ouabain | 10~7M ouabain | ||||
| Maximum
effect |
120 min after ouabain addition | Maximum
effect |
120 min after ouabain addition | ||
| 0-45 | – – 80-.:. 7 | + 97 H 5 | + 94 ± 6 | Sj|l57 ± 11 | + 150 ± 8 |
| 0-9 | – 55 ± 4 | .+ 118 ± 9 | + 114® 11 | + 139 ± 8 | + 90 ± 11 |
| 1-8 | 0 | 102 + 7 | + 100 Hs | -1- 146 ± 10 | 12 ± 8 irreg. |
| 3-6 | + 73 ± 8 | + 59 ± 4 | .+■ 51 ¿a 8 | + 87 S5 | H 33 ± 5 irreg. |
| 7-2 | + 84 ± 7 | + 24 ± 3 | + 18 IK 7 | I- 35 :!; 4 | — 66 ± 8 irreg. |
It has been stated that if digitalis glycosides act via a calcium effect then digitalis and calcium effects should be the same. The search of the literature has shown that digitalis and calcium manifestations on the heart show similarities and differences. A chemical agent may simulate another agent and may have actions of its own. Thus this line of comparison may not be too fruitful for an understanding of digitalis action. However, there are some remarkable similarities in action which have been studied. Thus neither calcium17 nor digitalis18 in a resonable dose range influenced the resting potential of cardiac muscle cells. Calcium had no striking effects on the transmembrane action potential17 nor did digitalis in therapeutic doses produce significant changes in the action potential.18 Toxic doses of digitalis eliminated the plateau and shortened the action potential.18 Both calcium and digitalis in toxic concentrations caused irregularities of the cardiac beat and these two substances acted sinergistically.15 Traut-
A. Farah and P. N. Witt
142
wein and Witt19 have shown that effects of low calcium on frog heart muscle action potentials can be reversed by ouabain. Contractility of cardiac muscle is improved by both substances and in the mammalian heart at least this is accomplished by increasing the rate of contraction without changing the length of the contraction and relaxation cycles. Calcium as well as digitalis inhibited the relaxing factor of cardiac muscle.20 Both substances eliminated the staircase phenomenon in amphibian cardiac muscle and produced a so-called reverse staircase.15 Both digitalis21 and calcium22 increased and later reduced the frequency-force relation in mammalian cardiac muscle and both first increased and then eliminated post-stimulation potentiation.21,23 The effects of digitalis are frequency-dependent.24-26 Recent unpublished studies by Teiger have shown that the speed of calcium effects is also frequency dependent.
Some dissimilarities are: the speed of action of calcium was much greater than that of digitalis. Cardiac glycosides reduced K+ influx in red blood cells27 and increased the rate of loss of K+ in cardiac28,29 and some striated muscle preparation.30,31 Ca++ did not change K+ influx in red blood cells32 nor did it increase K+ loss from striated muscle of the frog33 nor from the isolated mammalian heart.34,35 Increase in Ca++ markedly prolonged the decay of poststimulation potentiation (see Fig. 8), ouabain had no such effect.21 A decrease in extracellular sodium ion increased calcium effects on the heart.36,37 Unpublished observations from our laboratory have shown that a decrease of Na+ to 50% of normal practically eliminated the ouabain induced positive inotropic effect while increasing sodium to 125 or 150% of normal did not significantly modify this ouabain action (see Table III). Calcium decreased sodium permeability and was necessary for maintaining low intracellular sodium concentrations.34 Digitalis is claimed not to have this effect. However, in therapeutic doses digitalis glycosides reduced intracellular sodium concentration in rabbit heart muscle4 (see Table I) and in the human heart.6 The above differences make it unlikely that digitalis glycosides act by substituting for Ca++. However, the similarities do suggest that, in some subtle way, digitalis may act by modifying calcium activity.
Wilbrandt15 in a review published in 1958 has attempted to relate digitalis action to Na+, K+, and Ca++ transport. His hypo-
Cardiac glycosides and calcium
143
Table III
Effect of Na+ concentration in tyrode solution on the possitive inotropic of ouabain (2- 5 X 10~8 M)
Rabbit left ventricular preparations. Bath volume 100 ml. Temp. 37°C. In those solutions where NaCl was reduced sucrose was substituted in isosmotic amounts. Ventricular strips were stimulated 3/sec. After a control period of 30 min in normal Tyrode (control period) the ventricular strips were bathed in the experimental solution (experimental period). Ouabain was added 30 min after changing to experimental solution. Each value is the average ± S.E. obtained from 8-16 experiments.
| Na+ cone, in Tyrode solution (mM/1.) | Contractility dui ing experimental period (% of control) | Effect of ouabain (2-5 X 10“8M) on contractility as % of experimental contractility | |
| Maximum effect | 120 min after ouabain addition | ||
| 69-5 | §¡$-18 ± 5 | + 8 + 4 | ^H9-6 + 7 |
| 104-3 | + 31 ± 9 | +23K‘4 | -0-5 ± 2 |
| 139 | 0 | + 69» 4 | + 69-2 + 4 |
| 173-7 | ■ 40 + 7 | + 56 ±4 | + 53 ±4 |
| 208-4 | ~9±8 | 58 ± 7 | +53 í,-7 |
| Na+=69-5;Ca+ + *iO-45 | -44 ± 3 | + 9 + 3 | -18 +5 |
thesis was that the concentration of intracellular Ca++ ion was proportional to the amplitude of contraction. Any factor that increased intracellular Ca++ concentration directly or indirectly would increase contractility. Thus an increase in the extracellular calcium concentration would increase intracellular Ca*”4” and contractility. A competition between calcium and sodium ion at the membrane has been demonstrated by Wilbrandt and Koller36 and these observations have been confirmed and extended by Lüttgau and Niedergerke.37 Thus if Na+ accumulated intracellularly it would have competed with the transport of Ca++ out of the cell and would have caused Ca++ accumulation and a positive inotropic effect. Furthermore Na”‘” and Ca+ + supposedly competed for entry into the cell thus increased extracellular Na+ should have decreased while a decrease in Na+ should have increased Ca++ entry. Niedergerke38 has shown that a decrease in Na+ ion in the bathing fluid increased Ca45
A. Farah and P. N. Witt
144
uptake of heart muscle. Consequently a lowering of outside Na-1 or an increase in Ca++ concentration should cause increased cardiac contractility. This agrees with the observed facts. A decrease in extracellular K+ concentration increased contractility and increased intracellular sodium concentration probably because of the coupling of the transport of these two ions. An increase of intracellular Na+ should thus have decreased Ca++ out-flux and as a consequence intracellular Ca+4 and contractility should have increased. Niedergerke and Harris39 have shown that strontium-89 uptake of the frog heart was increased in K+ free solutions. As a consequence of digitalis action on Na+ and K+ transport, sodium should have accumulated intracellularly thus causing intracellular Ca++ accumulation and a positive inotropic effect. The finding that Ca4 1 accumulates intracellularly during digitalis action would thus be a crucial aspect of the Wil-brandt hypothesis for digitalis action. Wilbrandt and Caviezel40 have claimed that Ca45 efflux from heart muscle was reduced by cardiac glycosides. On the other hand Harvey and Daniel41 and Thomas et al.42 have shown that Ca45 uptake was not changed by non-toxic concentrations of cardiac glycosides. Unpub-blished experiments by Witt have shown that Ca45 uptake by rabbit auricles ‘stimulated at one per second was not increased by 10—7 M ouabain; a concentration that produced a marked positive inotropic effect (Fig. 2). Thomas et aZ.42 and Sekul and Holland42“ have shown that concentrations of ouabain which can produce a cardiac contracture increased Ca45 uptake. Similarly, Witt observed a significant increase in Ca45 uptake when a highly toxic concentration of ouabain (10~6 M) was used (Fig. 2). The data of Witt can be interpreted to mean that neither in- nor outflux of Ca45 was modified by 10~7 M ouabain since interference with either flux should have modified Ca45 uptake after prolonged exposure. The above data thus did not support the hypothesis of Wilbrandt and similar to K+ changes the cellular Ca++ concentration seems to be related to the toxic digitalis effects. Furthermore the hypothesis of Wilbrandt was based on the contention that positive inotropic effects of digitalis glycosides are causally related to the inhibition of K+ influx. The findings of Lee et al.3 and Tuttle et aZ.4 throw some doubt on this interpretation and thus the interrelations between Na+, Ca4 + and Kh fluxes and digitalis action discussed by Wilbrandt are probably
Cardiac glycosides and calcium
145
not causally related to the positive inotropic effect induced by cardiac glycosides.
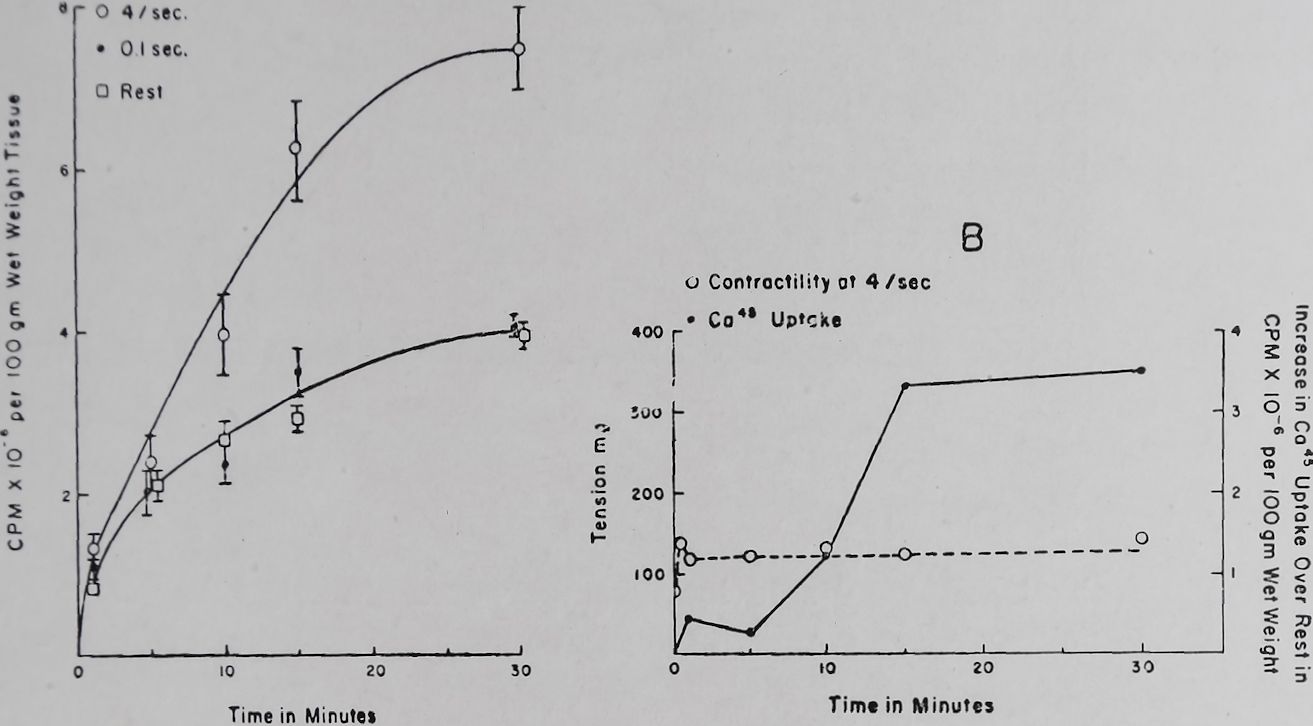
In rabbit auricles an increase of the external Ca++ concentration from 1*8 to 3-6 mM produced an increase in contractility of about 75’%> and was dependent on the rate of stimulation. At a rate of 4 per sec maximum effects on contractility were attained about 30 sec after addition of the Ca++ (unpublished). A rate of 4 per sec also increased Ca45 uptake (Fig. 3A). However, maximum calcium uptake was attained only after 30 min. Thirty seconds after increasing the calcium concentration uptake of Ca45 did not exceed more than 5% of the maximal calcium uptake (Fig. 3B). In auricles that had been exposed to a high Ca++ concentration containing Ca45, a return to a normal calcium concentration produced full reversal on contractility within 30 to 60 sec. Ca45 loss at this time was no more than 5-10°/o of the total calcium in the heart. It is thus clear that only a small fraction of the total Ca++ uptake could be related to the changes in contractility and the methods so far applied are not suitable for
Fig. 2. Effect of ouabain on the uptake of Ca45 by left auricles of the rabbit. Tyrode solution. Temperature 37°C. Bath volume 2-5 ml. Each column represents uptake of Ca4® 40 min after exposure to the radioactive Ca++ and the standard error. The values of Ca45 are given in arbitrary units. Each column represents the average uptake of 16-32 left auricles (unpublished data of Witt).

146 A. Farah and P. N. Witt
detecting relatively rapid and small changes in Ca”1“*’ uptake. The speed of action of Ca++ and the small quantity involved suggests that Ca++ effects on contractility are primarily due to changes on the membrane and are not likely, or only secondarily due to intracellular changes in calcium concentration.
A

Fig. 3. Effect of rate of stimulation on Cats uptake and contractility of rabbit left auricles. Temperature 37°C. Bath volume 100 ml. Tyrode solution. Calcium was increased from a normal of 1*8 to 3-6 mM per 1. (containing Ca4®). Ca4® uptake was determined at 1, 5,
15 and 30 min after the change to the higher Ca++ concentration. Contractility was recorded all through the experiment.
A. Ca45 uptake at stimulation rates of 0, 0-1 and 4 per sec.
B. Relation of Ca4® uptake and change in contractility at a stimulation rate of 4 per sec. Ca4® uptake plotted was the difference between the values obtained at rest and at 4 per sec. stimulation.
(Unpublished data of D. Teiger).
However, some evidence does point to a possible intracellular site of Ca++ action. Sodium fluoride had a positive inotropic effect on the isolated frog heart43’44 and on isolated rabbit auricular tissue45 stimulated at low rates. Clark46 and Loewi47 have shown that certain lipoids that can bind Ca++ also produced a positive inotropic effect in the frog heart. On the other hand Ca 1 ‘ chelators such as oxalate, citrate and ethylenediaminetetra-acetic acid reduced or stopped cardiac contractions possibly because of the formation of extracellular Ca++ complexes. During fluoroacetate poisoning citrate accumulated in rabbit auricles45
Cardiac glycosides and calcium
147
and contractility was significantly increased at low rates of stimulation.45 This suggests that penetration through the membrane may be an important factor in determining whether a Caf+ chelator produces a positive or negative inotropic effect. Thus citrate, when added to the bath, produced a negative, but when formed intracellularly it produced a positive inotropic effect. If the fluoroacetate effect was due to an increase in intracellular citrate then an increase in intracellular Ca+ + should be detected. Peters in his Croonian lecture48 states that in rat hearts poisoned with fluoroacetate no increase in cardiac Ca++ could be demonstrated. However, in hearts of Xenopus laevis a threefold increase in Ca++ could be shown 4 days after fluoroacetate poisoning. This suggests that intracellular citrate accumulation can produce an increase in the cardiac Ca++ concentration and this may be the cause of the increased cardiac contractility observed in certain fluoroacetate poisoned isolated heart preparations. The evidence presented is indirect and suggests that Ca++ chelators if they can penetrate the cell membrane produce a positive inotropic effect by virtue of an increase in intracellular Ca++ or by their ability to change intracellular calcium to some form which makes it available for the contractile mechanism. The influences of fluoride, fluoroacetate, the lipoids and the cardiotonic complex of Hajdu49 on digitalis action would thus be of interest and may assist in unraveling some of the complexities of digitalis effects.
Since the studies of Bowditch on the “Treppe” or staircase in the frog heart, a number of rate-dependent types of potentiation have been described (for a review see Mommaerts et al.50). It was of interest to study the effects of cardiac glycosides on these rate-dependent changes in contractility, especially so since Hajdu51 has shown that Treppe is eliminated by cardiac glycosides in the frog heart. Furchgott and DeGubaroff52 have observed that in the guinea pig isolated auricle k-strophanthin increased the contractility at low rates of stimulation and thus eliminated the frequency-force curve. Our own experiments were conducted mainly in rabbit left auricular preparations which were usually less than 0*5 mm in thickness and could be stimulated over a long period of time with relatively small changes in contractility. In this preparation a stepwise increase in rate from 0*1 to about 0-5 per sec produced a decrease in contractility, increasing the rate beyond 0*5 per sec caused an increase followed by
148
A. Farah and P. N. Witt
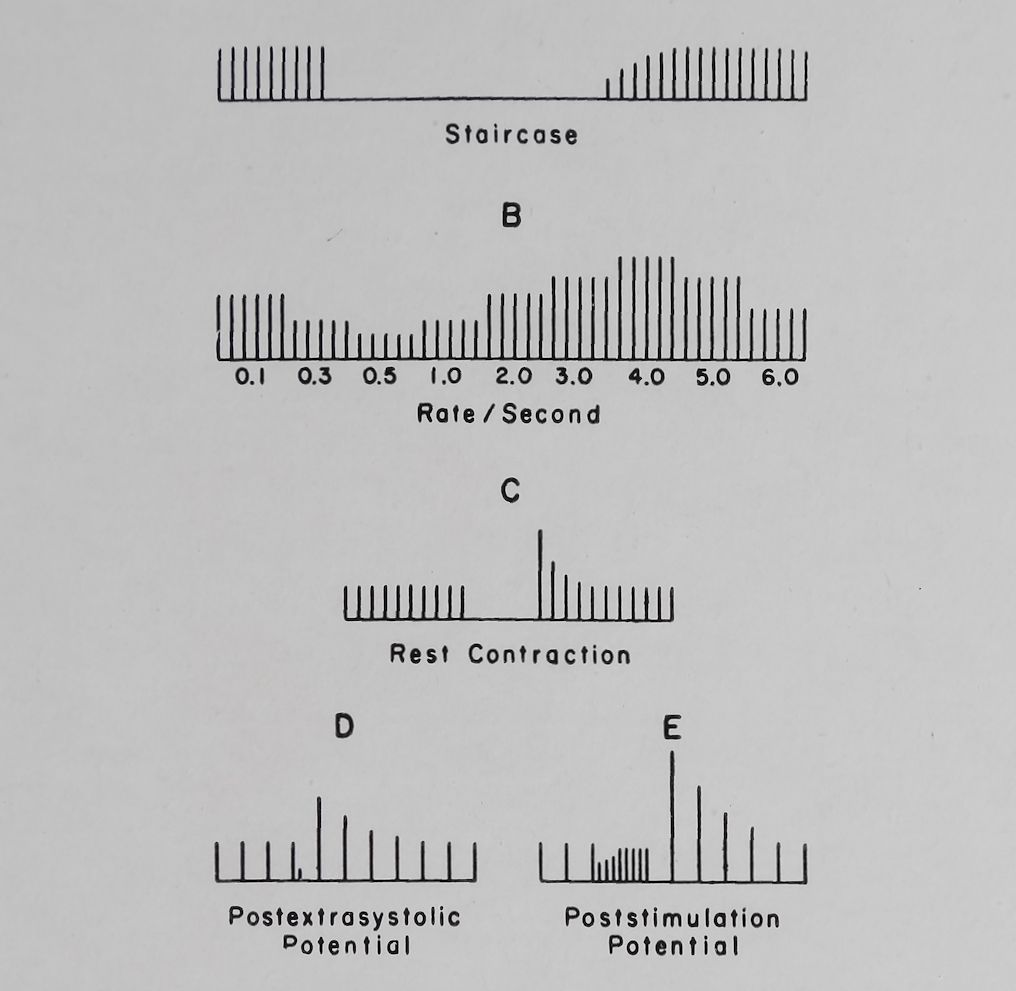
a second decline in contractile force (see Fig. 4). This frequency-force curve (S-shaped curve) has been described and analyzed by Kruta and co-workers53,64 and by Katzung et al.45
A
Fig. 4. A diagrammatic representation of: A. Treppe (staircase). B. Frequency-force relation. C. Rest contraction. D. Postextrasystolic potentiation and E. Post-stimulation potentiation.
Another type or potentiation, namely, “post-stimulation potentiation” (PSP) was first observed by Langendorff55 and was studied in some detail by Rosin and Farah.56 PSP was produced by interjecting a short period of rapid stimulation followed by a rest period before returning to the basal rate of stimulation. The first few beats after the episode of rapid stimulation were stronger than the basal beat and within 5-10 beats the tension developed returned to control levels (see Fig. 4). A number of factors which influenced PSP such as the rate and length of the rapid stimulation, intensity of stimulation, the length of the rest period and the effects of temperature have been described by Rosin and Farah60 and Katzung and Farah.57 Tuttle and Farah21 have recently studied the effects of ouabain and acetyl

Cardiac glycosides and calcium
149
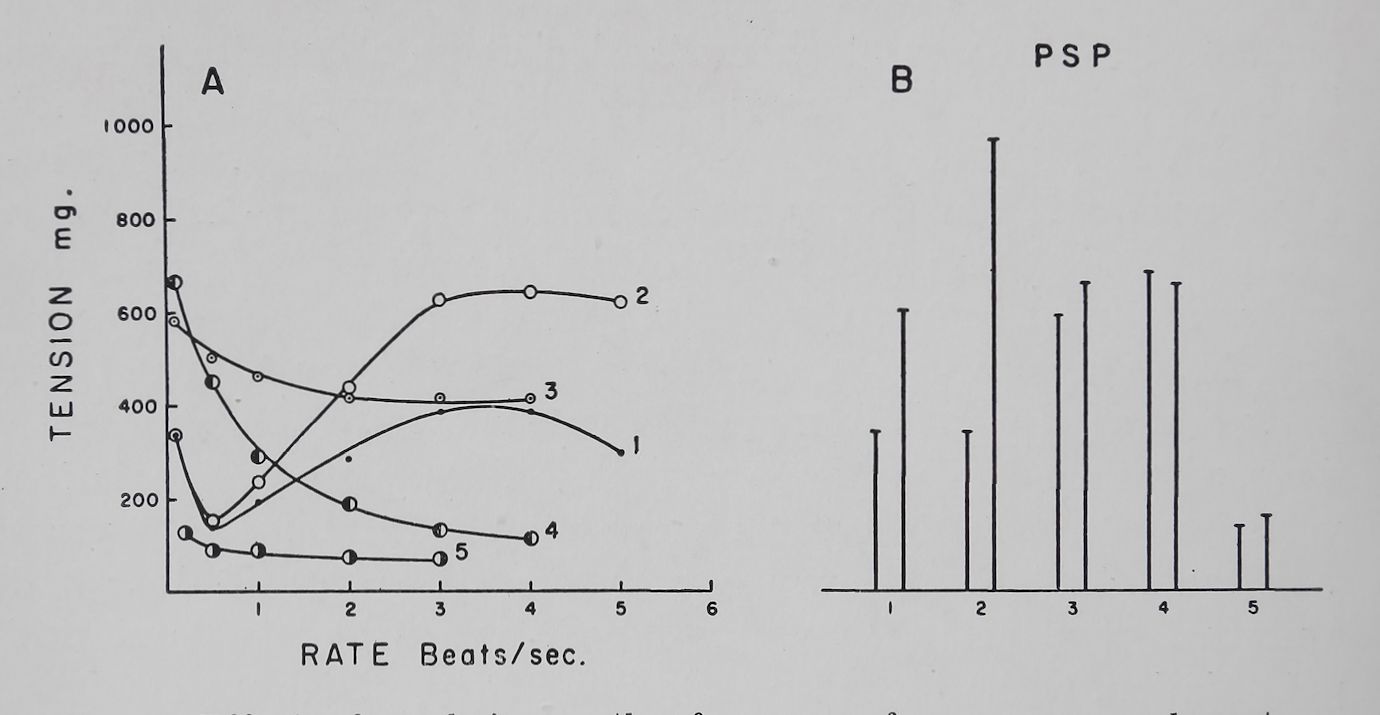
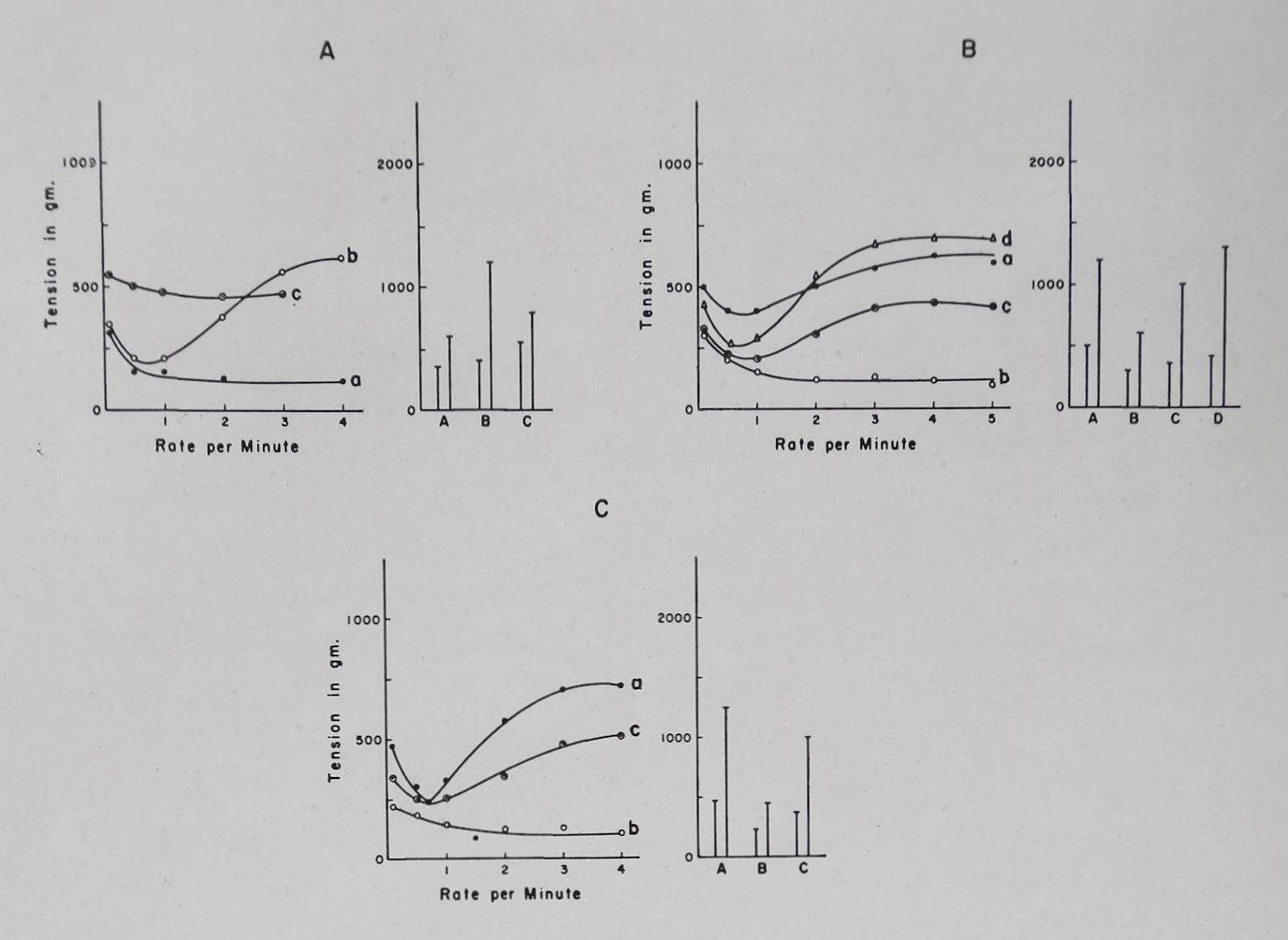
strophanthidin on the frequency-force curve and on post-stimulation potentiation. With a relatively low concentration of ouabain (2*5-5 X 10—8 M) the increased contractility was seen mainly at the high rates of stimulation. Higher concentrations (10 ‘7 — 10~6 M) of ouabain produced a second type of effect, namely an increase of contractility at the low rates of stimulation thus eliminating the frequency-force curve, followed by a decline in contractility at high rates and finally a marked loss of contractility at all rates. Concomitant with these changes, PSP was increased by the low concentration and reduced or eliminated by the higher concentration of ouabain (Fig. 5).
Fig. 5. Effect of ouabain on the. frequency-force curve and poststimulation potentiation. Rabbit left auricle. Temperature 37°C. Bath volume 50 ml. Tyrode solution.
A. Frequency-force curves.
B. Basal contractility and the first post-stimulation potentiation. Post-stimulation potentiations were determined at a basal rate of 0*1 and a fast rate of 6 per sec (for 5 sec) followed by a 10 sec
rest period.
(1) Control; (2) 45 min after 5 X 10~8 ouabain; (3) 50 min after 10~7 M ouabain; 4; 18 min after 2 X 10~6 M ouabain; (5) 33 min after 2 X 10—° M ouabain.
Similar results have been observed with acetyl strophanthidin. These observations have been extended to cat auricular muscle and rabbit and guinea pig ventricular muscle strips and the results were essentially the same. Changing the temperature from 37*5° to 27cC resulted in ouabain effects which were

150
A. Fahah and P. N. Witt
qualitatively the same as those observed at the higher temperature.21
The question arose whether some of these effects observed are due to toxic manifestation of the glycoside. A number of rabbits were given divided doses of ouabain until ventricular extrasys-tolies or ventricular tachycardia became apparent (total dose 0*15 mg/kg, 0*2 mg). The hearts were removed and isolated strips of the left auricle and right ventricle were compared with preparations obtained from rabbits receiving saline. In all these experiments a dose of ouabain which produced toxic manifestations in the intact rabbit eliminated the frequency-force curve and reduced post-stimulation potentiation. If 0-075 of ouabain per kg of rabbit was given in divided doses a definite increase in contractility of the ventricle could be recorded by in vivo methods. In all these preparations obtained from rabbits receiving a non-toxic but therapeutic dose, the frequency-force curve and post-stimulation potentiation were present in both auricular and ventricular strips. Comparison between the control groups and the ones receiving the therapeutic dose of ouabain did not show any significant differences.21 This was not surprising since the frequency-force curve and post-stimulation potentiation varied a great deal in control preparations. The important point was that rabbit hearts which received a therapeutic dose had a normally appearing frequency-force curve and post-stimulation potentiation, while those hearts which had received a toxic dose, showed a loss of the typical frequency-force curve and a reduction in post-stimulation potentiation. We thus feel justified to consider those concentrations of ouabain which produced an elimination of the frequency-force curve and a reduction in post-stimulation potentation as “toxic” doses and those concentrations that increased the frequency-force curve and poststimulation potentiation as “therapeutic” doses.
An occasional auricular or ventricular preparation showed a flat frequency-force curve and weak PSP. Addition of 2-5-5 X 10_8M ouabain to such preparations produced a reappearance of the frequency-force curve and an increase in the PSP. Raising the dose of ouabain to 10~6M caused a disappearance of the frequency-force curve and a reduction in PSP (Fig. 6A). Pentobarbital depressed cardiac contractility of auricular and ventricular strips, flattened the frequency-force curve and reduced
Cardiac glycosides and calcium
151

Fig. 6. Effect of ouabain on the frequency-force curve and poststimulation potentiation in isolated rabbit heart muscle strips. Tyrode solution. Temperature 37°C. Bath volume 50 or 100 ml. First column = basal contraction. Second column = post-stimulation contraction. Basal rate = 0*1 per sec; fast rate = 6 pet sec for 5 sec;
rest period 10 sec.
A. Effect of ouabain on a right ventricular strip of a rabbit which did not show a normal frequency response: (a) control curve;
(b) 55 min after addition of ouabain 5X10—8 M; (c) 27 min after
addition of ouabain 10 ~7 m
B. Effect of ouabain on pentobarbital depressed contractility of rabbit left auricle, (a) control curve; (b) 30 min after 1*5 mg°/o of pentobarbital in the bath; (c) effect of 10_8 M ouabain on the pentobarbital depressed auricle; (d) 48 min after 5 X 10—8 ouabain had
been added to the pentobarbital depressed auricle.
C. Effect of ouabain on a rabbit left auricle depressed by lowering Ca++ concentration: (a) control curve (Ca++ = 1*8 raM); (b) effect of 0-36 mM Ca++; (c) effect of ouabain (2*5 X 10—8M) on auricular
contractility depressed by a low Ca++ concentration.
PSP. Ouabain 10—8 — 5 X 10“8 M produced a positive inotropic effect especially at the high rates and restored the PSP to control values (Fig. 6B). In a similar fashion reduction in the calcium concentration from a normal of 1*8 to 0*8 mM per 1. produced
A. Farah and P. N. Witt
tsa
a reduction in the frequency-force curve and PSP which was partially restored by ouabain (Fig. 6C). These results show that one of the important actions of ouabain is the restoration of the frequency-force curve and the PSP and it is likely that this effect is the major therapeutic action of this cardiac glycoside.
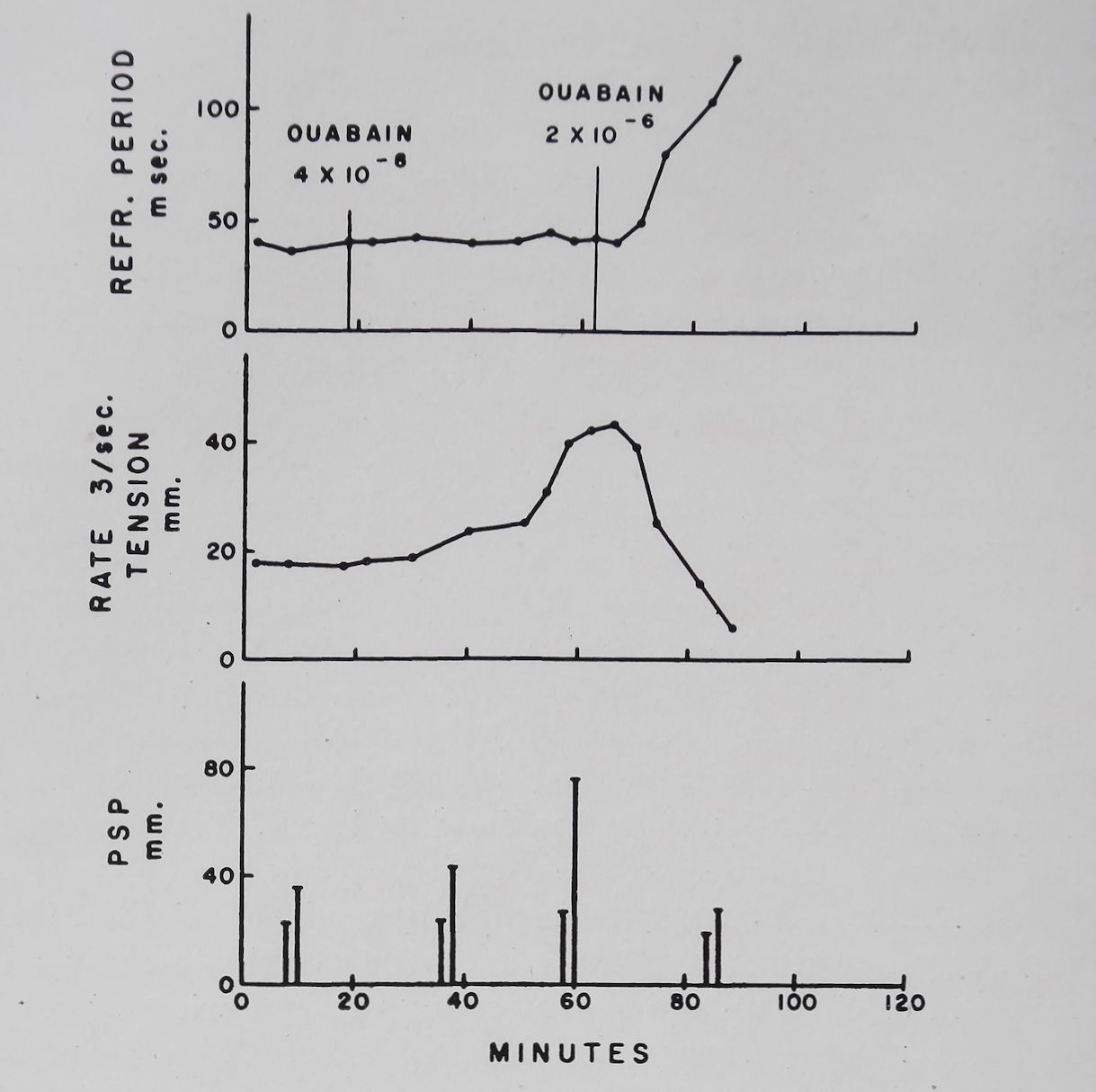
The cardiac glycosides in toxic concentrations eliminated the frequency-force relation and reduced PSP. It is known that Strophanthin can decrease the rate of rise of the action potential18,58 and can increase the refractory period and decrease excitability of auricular tissue.59’60 Furthermore Niedergerke61 has shown that calcium produced a reverse staircase in amphibian heart muscle in concentrations that reduced the duration and height of the action potential. These observations suggested the possibility that the decline in PSP and the frequency-force curve may be related to the electrical changes produced by ouabain. Fig. 7 shows the effects of therapeutic and toxic doses of ouabain on the refractory period, contractility, and PSP. It is apparent that with the therapeutic dose of ouabain (5 X 10~8M) contractility and PSP increased while the refractory period was unchanged. Increasing the ouabain concentration to a toxic level (2 X 10~6 M) produced a reduction in contractility and PSP and a significant increase in the refractory period. From the above discussion it is clear that digitalis glycosides influenced the frequency-force curve and PSP. For any further understanding of digitalis action it is thus imperative to inquire into the nature of the frequency-dependent inotropic effects.
A number of different types of frequency-dependent inotropic effects have been described and these include “Treppe”, the frequency-force relation, post-extrasystolic potentiation, the rest contraction in mammalian muscle, and post-stimulation potentiation (for a review see Mommaerts, et ai.).50 It is logical to assume that all these phenomena are closely related and it is our experience that factors which tend to modify one type of potentiation also modify the others in the same direction. Some of these phenomena occur in the intact heart in situ,62 thus making it likely that these potentiating mechanisms are inherent characteristics of heart muscle and are not an artifact due to isolation of the muscle. In the present discussion we will mainly restrict ourselves to an analysis of post-stimulation potentiation (PSP) since this has been investigated in greater detail in our
Cardiac glycosides and calcium
153
Fig. 7. Effect of ouabain on contractility, effective refractory period and post-stimulation potentiation in an isolated rabbit left auricle. Tyrode solution. Temperature 37°C. Bath volume 100 ml. Upper graph: effective refractory period determined at a rate of stimulation of 3 per sec. Middle graph: tension in mm per deflection measured at a rate of stimulation of 3 per sec. Bottom graph: PSP determined at a basal rate of 0-1 and a fast rate of 6 per sec (for 5 sec) and a rest period of 10 sec. (Unpublished experiment of
F. Scheider),
laboratory. Rosin and Farah56 have studied the PSP phenomenon and have shown that the length and the rate of rapid stimulation as well as the length of the rest period are factors which affect the extent of the PSP. They showed that an episode of rapid stimulation produced a PSP even if the rest period was extended to 5 min. Once stimulation was restarted the PSP declined exponentially and 50% decay occurred in 2-4 beats. The conclusions drawn were, that whatever caused the PSP was fairly

A. Fakah and P. N. Witt
154
stable and was utilized by each contraction in a constant proportion of the amount available. Rosin and Farah52 proposed the hypothesis that PSP was caused by a potentiating substance which was formed during each beat and was utilized by the next contraction. The higher the rate the more of this potentiating substance was produced. It has been suggested that “Treppe”, as observed in the amphibian heart, is due to a loss of calcium from heart muscle during periods of quiescence, and that calcium is restored to the muscle during activity.63,64 This hypothesis has more recently been expanded by Lüttgau and Niedergerke37 who proposed that a negatively charged Caf+ complex (RCa) is the “activator” of contraction. This “activator” is supposedly formed on the membrane or the endoplasmic reticulum and is moved during depolarization to some deeper portion of the muscle cell where it activates contractions by combining with actin or myosin or with the relaxing substance. In support of this hypothesis Niedergerke34 has shown that KC1 induced depolarization increased Ca45 uptake by the frog heart. Electrical stimulation has been reported to increase or not to change Ca45 uptake. Thus Niedergerke,65 Henrotte et al.66 and Witt67 using relatively low rates of stimulation were unable to show an increase in calcium uptake above resting values during stimulation of heart muscle. Shanes and Bianchi68 and Cosmos66 reported that in skeletal muscle, stimulation (60/min) increased Ca45 uptake above resting values. Sekul and Holland70 reported that stimulation rates of 20 per sec increased Ca45 uptake abovq resting values. Recent preliminary findings from our laboratory (D. Teiger, unpublished) have shown that with a stimulation rate of 0 • 1 per sec Ca45 uptake was not changed while at a rate of 4 per sec Ca45 uptake was significantly increased above resting values. These findings suggest that Ca++uptake is a rate-dependent phenomenon and may possibly clarify the discrepancies in the data that have been published previously. Niedergerke38 has also shown that a reduction of the outside Na+ concentration increased Ca45 uptake and the contractility of the isolated frog heart. All these findings on Ca++ uptake by the heart are compatible with the hypothesis of Lüttgau and Niedergerke that the activator is a calcium complex. A possible relation between the negatively charged “activator” of Lüttgau and Niedergerke and the potentiating substance of Rosin and Farah will now
Cardiac glycosides and calcium
155
be considered. The importance of external concentrations of calcium, sodium and potassium ion on “Treppe’’ have been documented by Niedergerke71 and Wilbrandt.15 A brief description of changes in external ion concentrations on PSP will be presented.
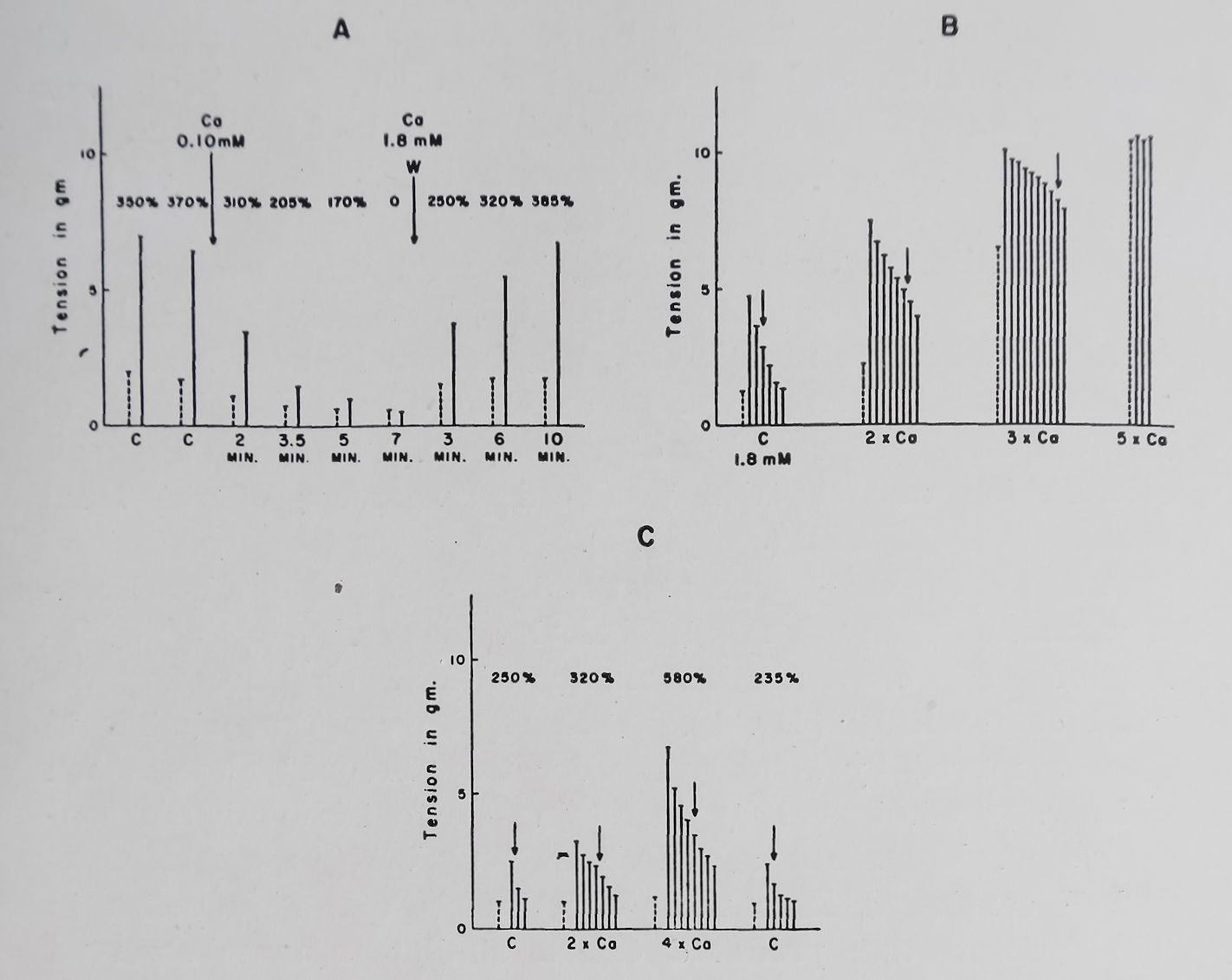
A reduction in Ca++ concentration from the normal one of Tyrode solution (1-8 mM per 1.) to 0*112 mM or zero produced in rabbit auricular muscle first a reduction in both basal contractions and PSP; this was followed by a further reduction of PSP and finally its complete disappearance. A return to a normal calcium ion concentration produced a recovery of the basal contractions and PSP (see Fig. 8).
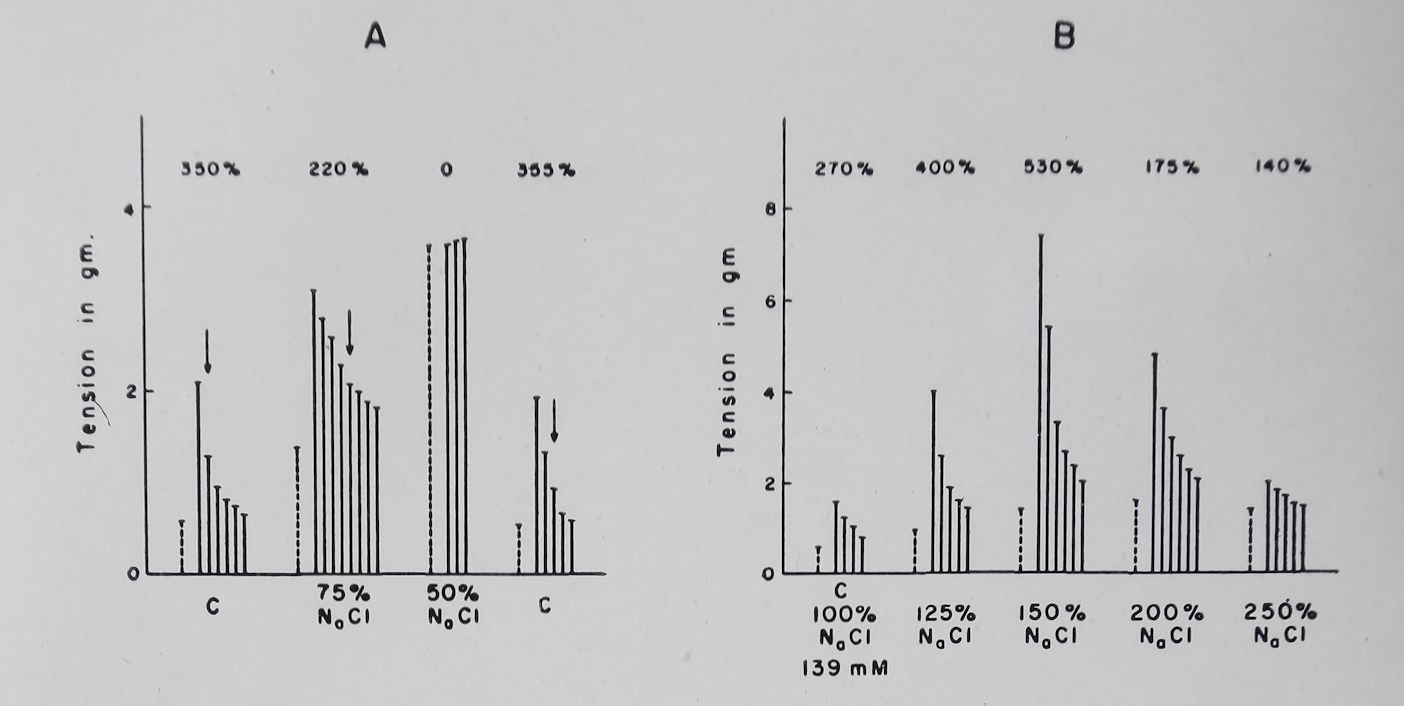
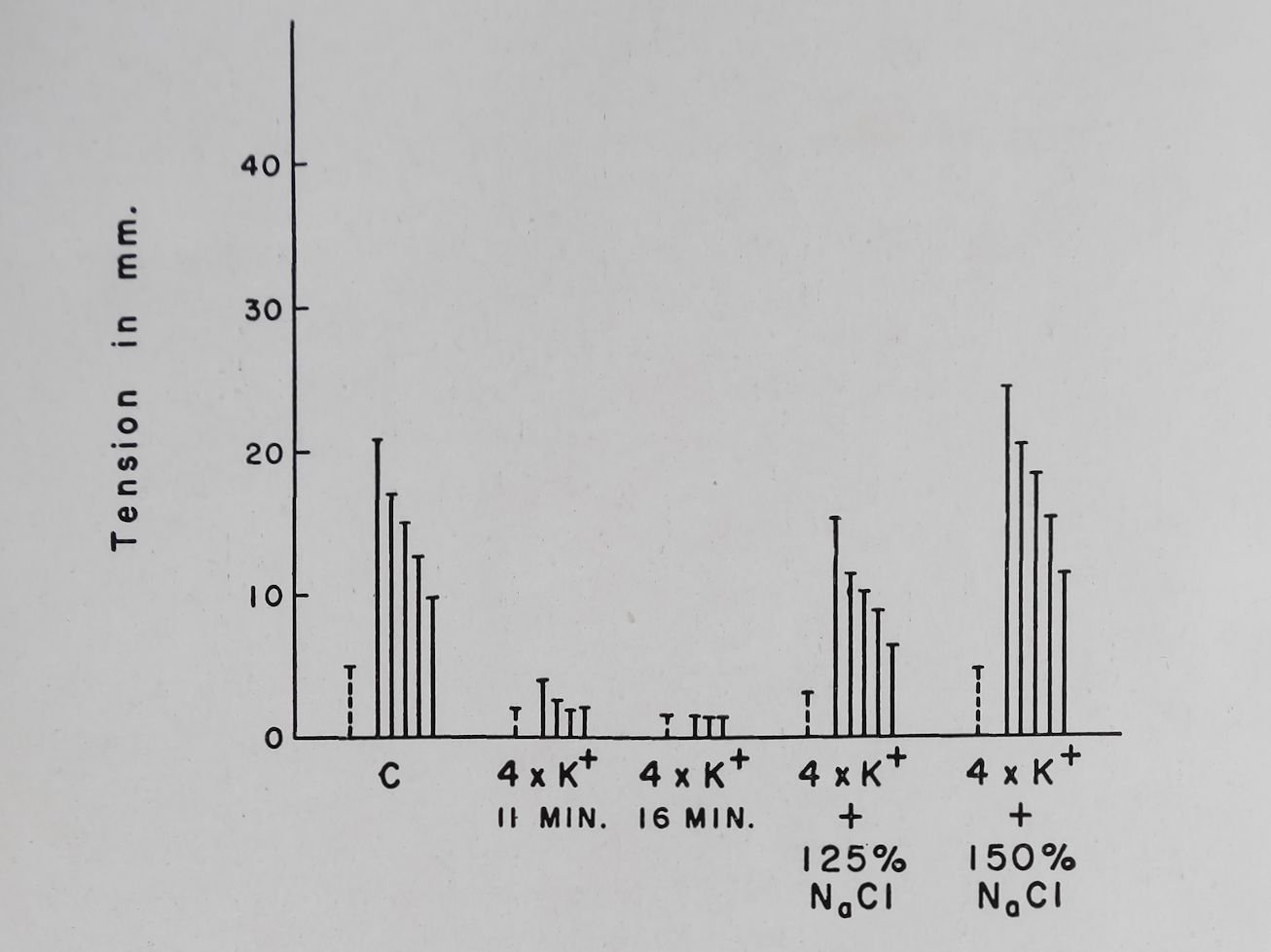
Doubling the Ca++ concentration increased both the basal contractions and PSP of rabbit auricular muscle. A further doubling of Ca++ concentration eliminated PSP by increasing the basal contractility (Fig. 8). These results are thus similar to those observed by Niedergerke for the “Treppe” phenomenon which was eliminated by increasing the outside calcium concentration. Wilbrandt and Koller36 and Lüttgau and Niedergerke37 concluded from their experiments that a reduction in Na+ concentration produced effects similar to those observed with an increase in Ca++. Lowering Na+ concentration up to 50% of normal produced effects on PSP similar to those observed with increased Ca++ (Fig. 9). Lowering Na+ concentration to 25-30% of normal (NaCl was replaced by isosmotic amounts of sucrose) produced a sharp reduction in the PSP and basal contractility and at these low concentrations PSP was completely absent. A striking effect of increasing Ca++ or lowering Na+ concentration was observed on the decay of PSP of rabbit auricular muscle. With normal Tyrode’s solution 50% decay of PSP occurred within 2-3 beats while with increased Ca+ or decreased Na+ the decay of PSP was markedly prolonged (Fig. 8 and 9).
Increasing Na+ concentration produced effects which depended on the amount of Na,+ added and on the K+ present in solution. With Tyrode’s solution containing 2*7 mM K+ increasing Na+ concentration by 25-100% above normal (139 mM/1) reduced or did not change the PSP. However, if the K+ ion concentration of the bathing solution was increased to 5*4 or 8*1 mM, an increase in Na+ ion concentration to 50 or 100% above normal
A. Farah and P. N. Witt
156
Fig. 8. Effect of changes in Ca++ concentration on basal and post-stimulation contractions. Stippled column = basal contraction. Continuous column = consecutive post-stimulation contractions. Basal rate 0*1 per sec; fast rate 6 per sec for 2 or 5 sec; rest period 10 sec. Numbers on top are the PSP in per cent of the basal contraction. Tyrode solution. Temperature 37°C. Bath volume 100 ml. C = control (1*8 mM Ca++). W — wash. Arrows indicate the 50°/o
decay of PSP.
A. Effect of reducing Ca+ + concentration on basal and post-stimula
tion contractions in the isolated rabbit left auricle.
B. Effect of increasing Ca++ on basal and post-stimulation
contractions in the isolated rabbit left auricle.
C. Effect of increasing Ca++ concentration in the isolated rabbit
right ventricular strip. (Unpublished experiments).
produced a reversal of the K+ induced depression of PSP (Fig. 9 and 10). Concomitant with the changes in PSP, Nat addition reversed the K+ induced increase in effective refractory period. Addition of sucrose or choline in isosmotic amounts did not produce these effects observed with sodium chloride. These

Cardiac glycosides and calcium
157
Fig. 9. Effect of changes in sodium chloride concentration on poststimulation potentiation. Rabbit left auricles. Tyrode solution. Temperature ST’C. Bath volume 50 ml. Stippled columns = tension produced by basal contraction. Continuous columns* consecutive poststimulation contractions. Arrows indicate 50°/o decay of post-stimulation potentiation. Numbers on top are the first PSP expressed in per cent of the basal contraction.
A. Effect of decreasing NaCl concentration. Normal Tyrode.
B. Effect of increasing NaCl concentration. In this experiment K+ concentration in Tyrode was 5*4 mM. PSP was produced at a basal rate of 0*1 per sec, fast rate = 6 per sec for 5 sec; rest period
= 10 sec (Unpublished experiment).
findings thus indicate that sodium ion counteracts the effect of K+ on both the PSP and the refractory period.
The data presented have shown that Ca++ is essential for the production of PSP. It is clear that complex ionic interrelations play a role in this phenomenon which qualitatively at least resembled those observed by Niedergerke61 and Wilbrandt15 for the “Treppe” phenomenon in the amphibian heart.
The potentiating substance postulated by Rosin and Farah may be identical with the “activator” proposed by Lüttgau and Niedergerke. The evidence presented is compatible with this hypothesis; however, it is by no means clear what the “activator” consists of except that it probably contains calcium ion. In support of this is the observation that only high rates of stimulation increase Ca45uptake (Fig. 3a) and it is only with episodes of high rates of stimulation that the PSP can be produced. Thus increasing Ca++ concentration or reducing sodium ion

158
A. Farah and P. N. Witt
Refr. Period 45 90 100 70 40 msec.
Fig. 10. Depression of PSP by an increase in the potassium concentration and its reversal by the addition of sodium chloride in the rabbit left auricle. Stippled column = height of basal contraction. Continuous column = Consecutive post-stimulation contractions. Ty-rode solution. Temperature 37°C. Bath volume 50 ml. Basal rate 0-1 per sec; fast rate = 6 per sec; rest period after rapid stimulation = 10 sec. Sodium chloride added was 25 and 50Vo above control values (139 mM). Refr. period = effective refractory period in milliseconds. (Data of F. Scheider).
concentration should increase the activator at the membrane. Na+ and K+ may possibly inhibit the “activator” by producing RNa+ or RK+ which are inactive and compete with Ca++ for the prosthetic group of the activator (R).
Niedergerke and Harris39 have shown that Ca45 uptake is increased by KCl-induced depolarization and the studies of Teiger alluded to previously have shown that frequent electrical depolarization also increased Ca45 uptake. These findings suggest that depolarization also increased Ca45 uptake and somehow caused the movement of Cafjj or the “activator” from the cell membrane to the interior of the cell. It is known that electrical

Cardiac glycosides and calcium
159
depolarization causes a sudden increase in membrane permeability and an influx of sodium ion. The rate of rise of the transmembrane action potential has been related to this increase in Na+permeability.73 It is thus possible that the sudden increase in sodium entry may be the factor that released the “activator” and moved it from the cell membrane to the interior of the cell. If the “activator” is the same as the “potentiating substance” then the reduction of the rate of sodium entry should have decreased the1 PSP. A decrease of sodium ion below 50% of normal reduced the basal contractility and eliminated PSP completely. Such a reduction in Na+ concentration eliminated the overshoot of the cardiac action potential73 and probably reduced the rate of entry of Na+ion during activity. Increasing K+ should reduce the resting potential.74 It is known that the rate of rise of the action potential is directly related to the resting potential. Thus an increase in outside K+ concentration should decrease the rate of rise of the AP and also the rate of entry of sodium ion and consequently reduce the PSP. Increasing the outside Na+ concentration should overcome these K+ induced manifestations and increase the K+ depressed PSP. The experimental facts presented support this hypothesis. West and Amory75 have shown that in isolated rabbit auricular muscle rates of about 4 per sec or , above decreased the rate of rise of the action potential. This can be correlated with the observation that the frequency-force curve in the rabbit auricle was usually depressed at rates greater than 4 or 5 per sec.
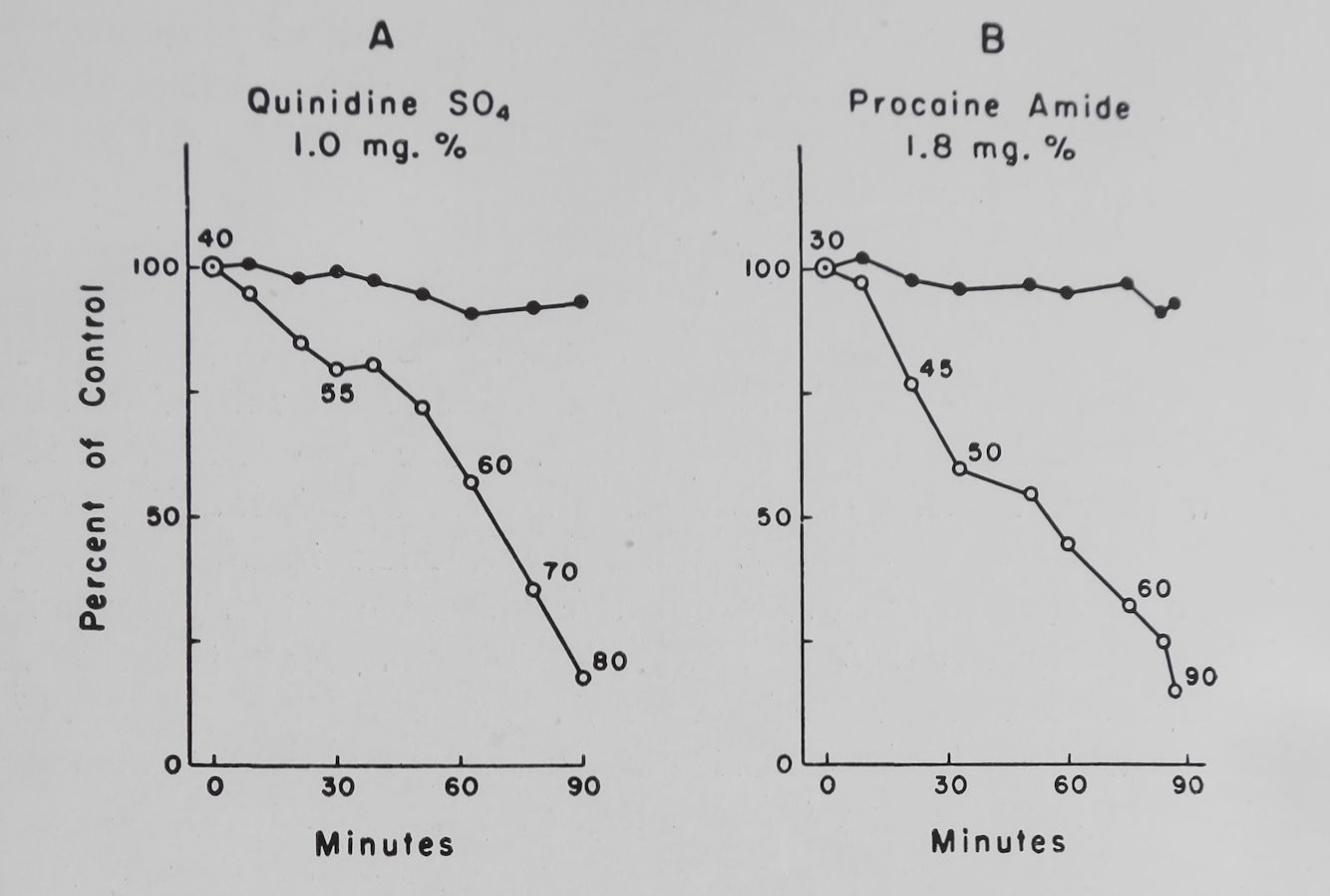
Quinidine and local anesthetics decreased the rate of rise and the overshoot of the action potential.17 With relatively low concentrations of quinidine the rate of rise of the action potential was reduced at the higher rates of stimulation.75 Thus according to the hypothesis presented quinidine and procaine amide should reduce the rate of entry of sodium, the frequency-force curve and PSP. The experimental observations agree with the hypothesis. Figure 11 shows that both quinidine and procaine amide in concentrations which prolonged the effective refractory period markedly decreased the PSP with relatively little change in the basal contractions. It has been postulated that an “available sodium carrier” is activated by depolarization to an “active sodium carrier”; this is then changed to an “inactivated sodium carrier”. During recovery the inactive sodium carrier is trans-
160
A. Farah and P. N. Witt
Fig. 11. Effect of quinidine sulfate and procaine amide on the basal contractility, post-stimulation potentiation and effective refractory period of rabbit left auricles. Normal Tyrode. Temperature 37°C. Bath volume 50 ml. Ordinate = basal and post-stimulation contraction (PSP minus basal contraction) in per cent of control values. Abscissa = time in minutes. Numbers along graph are the effective refractory period in milliseconds. Drug added at zero time.
(Unpublished data of Katzung and Scheider).
formed with time to the available sodium carrier.76 The time needed for this latter transformation may probably be related to the effective refractory period. Thus measurements of the effective refractory period will indirectly give us some information concerning the sodium carrier.
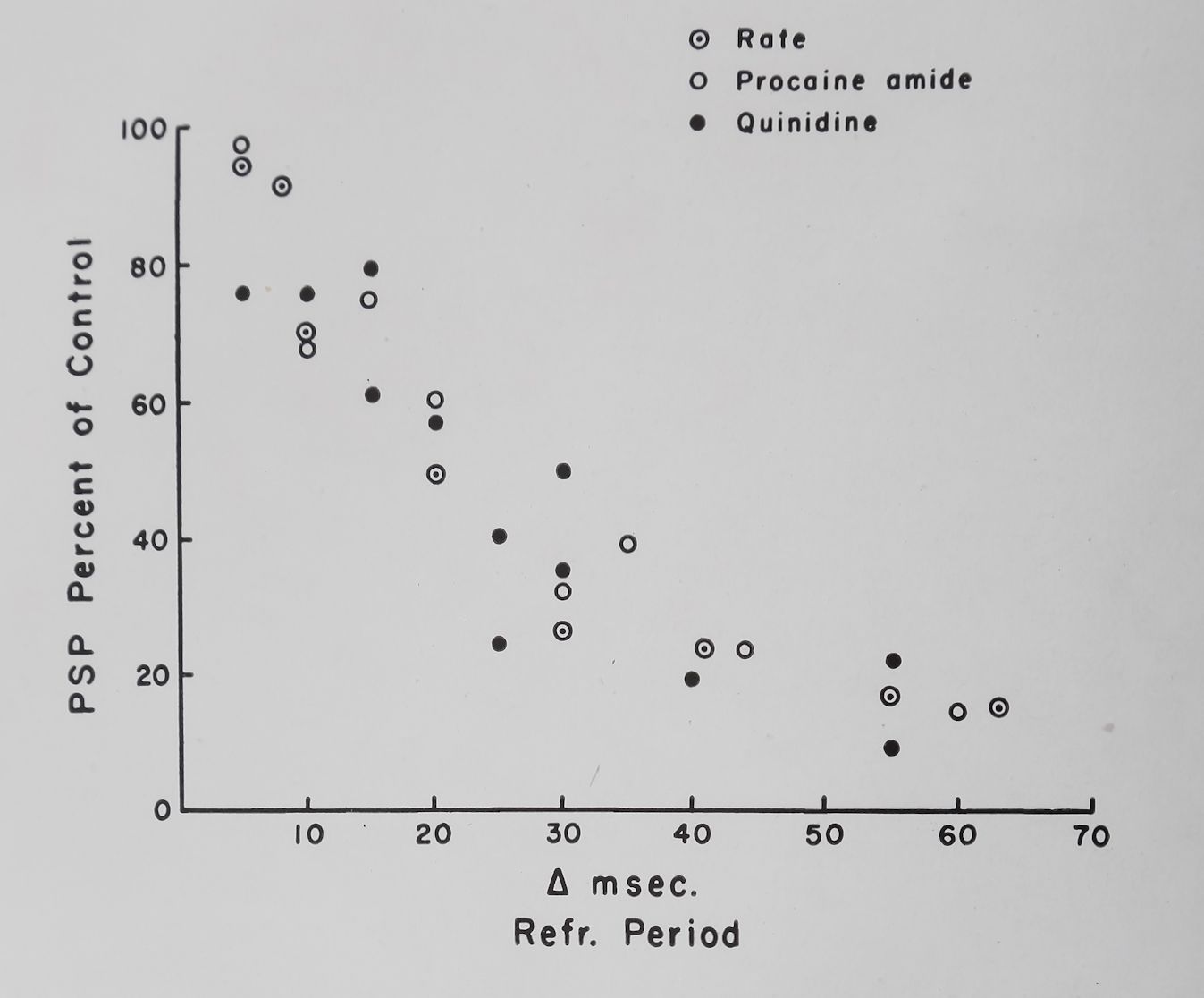
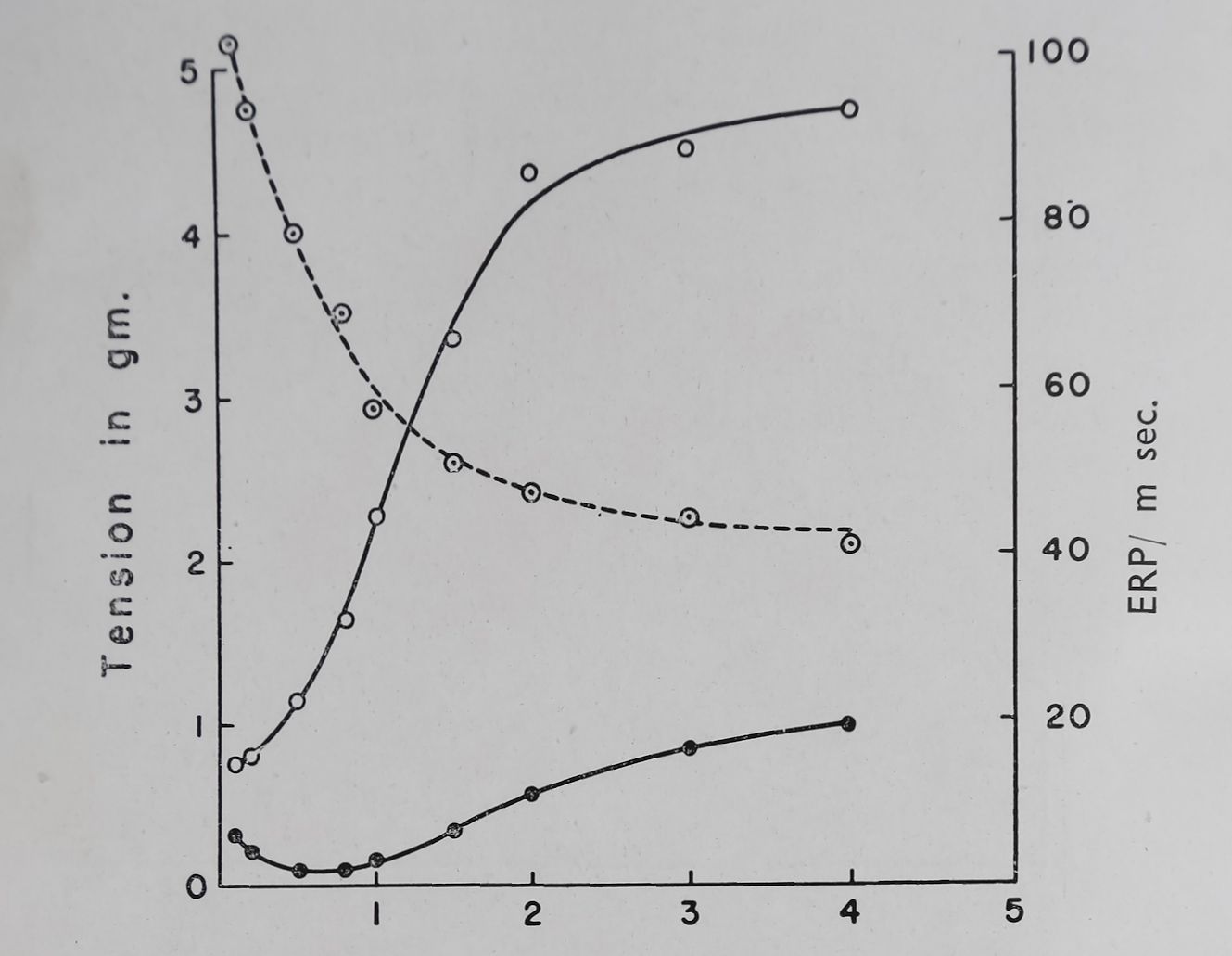
In Fig. 12 we have plotted changes in effective refractory period produced by quinidine and procaine amide against the depression of the PSP and a good correlation is quite apparent. If the rate of stimulation was increased the effective refractory period was decreased and PSP was increased. The average values obtained from five rabbit auricles are given in Fig. 13 and if these data are plotted in the same way as the quinidine and procaine amide data the correlation between refractory period changes and PSP is striking (Fig. 12). The conclusions drawn from all these experiments are that PSP is probably related to

Cardiac glycosides and calcium
161
Fig. 12. Relation of refractory period to post-stimulation potentiation (PSP). Ordinate = PSP minus basal contraction as per cent of control. Abscissa = change in effective refractory period produced by various procedures in milliseconds. For the rate changes the values of PSP obtained at a stimulation rate of 4 or 5 per sec were considered to be 100«/». (Unpublished data of Katzung and Scheider).
the calcium containing activator and Na+ entry during activity may play an important role in the production of some of these rate-dependent inotropic phenomena. Rosin and Farah56and Trautwein and Dudel77 have shown that during PSP neither the surface or transmembrane potentials were significantly changed from the control values. In many instances it was possible to produce a change in the contractility of cardiac muscle during high rates with no change of contractility at low rates. This suggests that the contractile mechanism per se is not involved in this potentiating mechanism and it is thus possible that PSP may be an index of the coupling mechanism.

Rate per Second
Fig. 13. Effect of rate of stimulation on basal contraction (•); poststimulation potentiation (O) and effective refractory period (O) in rabbit left auricles. Temperature 37°C. Bath volume 50 ml, (average of 5 auricles). Ordinate left = tension produced in grams. Ordinate right = effective refractory period in milliseconds. Abscissa = rate of stimulation per sec. (Unpublished data of D. Teiger).
In the isolated frog heart bathed in a normal Ringer solution the positive inotropic effect of cardiac glycosides is difficult to demonstrate. By reducing the Ca++ in the Ringer solution the inotropic effect of these glycosides became apparent.15 The data of Rohrer quoted by Wilbrandt15 show that maximal contractility was attained with the normal Ca++ concentration in Ringer solution. In rabbit isolated cardiac muscle bathed in normal Tyrode solution and containing 1*8 mM of Ca++ the positive inotropic effect of ouabain was easily demonstrated. In these preparations the maximal contractions were attained only when Ca++ had been increased to 3*6 or 7-2 mM. With high Ca1 + concentrations the inotropic ouabain effect was markedly reduced or eliminated (Table II). This observation confirms the finding

Cardiac glycosides and calcium
163
of Lee et al.3 and suggests that once a maximal calcium induced effect has been attained ouabain can show no further inotropic effects. This could be due to a specific effect of Ca++ or due to the attainment of maximal contractility. In our experiments ouabain was added 30 min after the change to the high calcium concentration. At that time contractility was about 50% of the maximum attained immediately after Ca++ addition. Thus it seems that the inhibition of the ouabain induced inotropic effect was due to the high concentration of calcium and not due to attainment of maximal contractility. Similarly Sanyal and Saunders78 have shown that the ouabain induced percentage increase in contractility in guinea pig ventricular strips was not influenced by the control tension since the same increase in contractility was observed in both the fresh and in the hypo-dynamic preparations. These points certainly require further study and are amenable to an experimental analysis.
The effects of lowering Na+ concentration on the inotropic action of ouabain are presented in Table III. It is clear that a substitution of sucrose for Naftj markedly depressed the inotropic effect of 2-5 X 10“3 M ouabain. The mechanism of this effect could be the relative increase in Ca++ concentration produced by the reduction in Na+, thus producing effects similar to those seen with an absolute increase in Ca++ concentration. However, if concomitant with the decrease in Na+, Ca++ was also reduced the contractility of rabbit ventricular strips was decreased but still showed a poor positive inotropic effect as a result of the addition of 2*5 X 10“8 M ouabain. This suggests that lowering the extracellular Na+ concentration might have effects on digitalis independent of the changes in relative Ca+‘1‘ concentration.
According to Caviezel and Wilbrandt79 low K+ increased while high K+ concentrations reduced the positive inotropic effect of strophantosid in the isolated frog heart. The findings of Lee et al.3on cat papillary muscles do not confirm these findings since K+ free solutions inhibited while high K4 (24 mM) if anything increased the per cent change in contractility induced by 1*37 X 10“6M ouabain. Preliminary results obtained in our laboratory indicate that 2*5 X 10~8 M ouabain did not produce a positive inotropic action when rabbit ventricular strips were exposed to a virtually K+ free solution. While in
164
A. Farah and P. N. Witt
the presence of 2*7 or 5*4 mM of K+ the same concentration of ouabain produced a 70-80 per cent increase in contractility.
Recently Lee20 has made the interesting observation that ouabain (10~® g/1.) could increase the adenosine triphosphate (ATP) induced contraction of glycerol extracted heart muscle fibers in the presence of the relaxing factor. A relation between Ca++ and ouabain was demonstrated since muscle fibers that did not respond to Ca++ did not respond to ouabain addition. Furthermore the disappearance of Ca++ was accompanied by the disappearance, of the relaxing effect of phosphocreatine (PC) (used to produce relaxing factor) thus linking the relaxing effect of PC with the stimulating effects of Ca++ and ouabain.
Benforado80 demonstrated that the rat heart showed only a negative type of frequency-force curve, that is, an increase in rate only produced a reduction in the contractile force. The rat heart is notoriously resistant to the action of cardiac glycosides but not to Ca++34 and it would be of interest if this correlation could be extended to other digitalis resistant species such as the mouse and toad. The experiments of Taeschler and Weidmann81 have shown that a reduction in temperature from 37 to about 18-19°C eliminated the positive inotropic effect of digitalis and Katzung and Farah57 have observed that in the turtle heart lowering the temperature caused the positive frequency-force curve to change to a negative one and the decay time of the PSP was markedly decreased. All these observations suggest that digitalis action and the mechanism responsible for the rate-dependent inotropic phenomena are related and that Ca++ plays an important role in these effects.
CONCLUSION
In this discussion we have attempted to review the relation of Ca++ to the positive inotropic effect induced by cardiac glycosides. The importance of dosage or concentration of these glycosides for any correlations between contractility and biochemical changes have been stressed. The changes in intracellular K+ are most likely not causally related to the positive inotropic effect but may be related to severe toxic manifestations produced by th ese glycosides.
The evidence presented shows that a major component of the positive inotropic effect of cardiac glycosides is related to an
Cardiac glycosides and calcium
165
increase in the frequency-force response and post-stimulation potentiation. The postulated “activator” of Lüttgau and Nieder-gerke and the “potentiating substance” of Rosin and Farah can be considered to be the same. It is postulated that the activator is a calcium complex formed on the membrane or endoplasmic reticulum. The rate of Na+ entry during a depolarization is probably related to a movement of the activator to deeper portions of the cell. The activator in some way combines with the contractile proteins or the relaxing factor, or both, and initiates the chain of events resulting in a shortening or sliding of the contractile elements. The activator is thus made available during each depolarization and utilized by the following contractions. Frequent depolarizations result in an excess of activator inside the cell thus the following contractions increase. This would explain both the Treppe and post-stimulation potentiation. According to this hypothesis the “activator” or the “potentiating substance” would be the link between the membrane phenomena and the actual contraction. If this should be a correct assumption then it will be apparent that poststimulation potentiation is a quantitative index of this coupling mechanism and that digitalis produces its major therapeutic effect via this coupling mechanism.
There are at least three probable sites which require Ca++ for a normal contraction. The first site is probably the cell membrane and here the presence of Ca++ is necessary for its normal excitability. The excitability of nerve82 and cardiac membrane83’84 are rather resistant to Ca++ deprivation although they can not function in the virtual absence of Ca++. Thus the action potential of the heart is relatively normal in the presence of Ca++ concentrations which do not support propagated contractions.
A second site, namely, the mechanism controlling propagated contractions of heart muscle is highly sensitive to variations in Ca++ concentration and it is likely that Ca++ is needed for a process which links the electrical manifestations to the contractile process (the coupling mechanism). This is the mechanism where the “activator” of Lüttgau and Niedergerke or the “potentiating substance” of Rosin and Farah seem to fit in.
A third possible site is the production of contractures, the extent of which are possibly related to Ca++ concentrations
A. Fahah and P. N. Witt
166
inside the cell and may be closely related to Ca++ bound to myofibrils or the relaxing factor of Marsh85 and Bendall.80 This area of muscle physiology, namely, the relation of Ca++to myofibrillar adenosine triphosphatase activity, the relation of the relaxing factor to Ca++, and the production of a soluble relaxing factor from microsomes have opened up new insights into the process of muscle contraction. It should be kept in mind that these studies on glycerinated fibers can not be related to the coupling mechanism since in these fibers the membrane has been virtually destroyed. It is likely that the contractions observed in these model systems are more closely related to those phenomena known as contracture rather than to the propagated twitch.
It has been previously proposed that the relaxing factor removes Ca++ from the myofibrils thus causing relaxation of these fibers. This was based on the observations that Ca++ inhibits the relaxing factor; that ethylenediamine tetra-acetate (EDTA) produced an inhibition of the myofibrillar adenosine triphosphatase and reversed the ATP induced contracture of glycerinated muscle fibers; that the binding of Ca++ to microsomal granules requires ATP and that the myofibrillar ATPase activity is dependent on the presence of small amounts of Ca++. However, Bozler87–88 has shown that EDTA removed only about half the amount of the Ca++ bound to myofibrils when it produced its relaxor effect and the remaining Ca++ (not removed) was sufficient to allow the fibers to contract on the addition of ATP. He thus interpreted relaxation in terms of a combination of the relaxor substance or EDTA with sites on the myofibril containing firmly bound Ca++. More recently Parker and Gergely89 have shown that EDTA or relaxing factor producing granules do not remove a Ca++ fraction from myofibrils essential for ATPase activity and that the relaxing activity is most likely due to the binding of the relaxing substance or EDTA to the contractile proteins.
The effects of Ca++ on the relaxing factor suggest that the soluble relaxing factor is probably inhibited by Ca++ or CaATP. The granules which produce the soluble relaxing factor combine with Ca (ATP is required for this combination) and maintain conditions favorable for the activity of the relaxing substance.89
Cardiac glycosides and calcium
167
That digitalis can produce permeability changes in the membrane is well established by the findings that cardiac glycosides in toxic concentrations can decrease excitability, increase the refractory period, decrease the rate of rise, and eliminate the plateau of the action potential. However, it is most likely that all these membrane changes induced by the cardiac glycosides are not causally related to the positive inotropic effects but are definitely related to the so-called toxic stage of their action.
The evidence presented here on the effects of low or “therapeutic” concentrations of cardiac glycosides on the rate-dependent potentiation phenomena (Treppe, frequency-force relation, and post-stimulation potentiation) suggest that the positive inotropic effect of these glycosides is related to that mechanism which couples the membrane to the contractile phenomena. The relation of Ca++ to this coupling mechanism has been discussed. It is likely that digitalis acts by sensitizing this mechanism to Ca++. If Ca++ has produced maximal effects (by increasing Ca++concentration in the bathing fluid) then digitalis produces no further positive inotropic effect. This is probably due to a saturation of some site by the high Ca++ concentration and not due to the attainment of maximal contractility.
The findings with reduced Na+ ion which inhibited ouabain action could also be explained on the basis of a saturation of a receptor by Ca++ since a reduction in outside Na+ increased Ca++uptake by the heart. The lack of action of “therapeutic” concentrations of ouabain in the virtual absence of Kf in the Tyrode solution could be explained on this basis since K+ removal from the Ringer’s solution increased Ca*’*”’” uptake by the heart. All these findings suggest that in some manner digitalis glycosides increased Ca++ activity possibly by promoting the formation of the “activator” but without increasing the total Ca++ content of the heart. It may be that digitalis can increase the concentration of the prosthetic group of the activator and thus more activator can be produced in the presence of the same Ca++ concentration.
However, the experiment with low Na+ and low Ca++ where the ouabain induced positive inotropic effect was inhibited suggests that Na+ entry during activity may in some way be involved in the increased contractility produced by ouabain.
168 A. Far ah and P. N. Witt
This aspect of the problem requires more experimental data before it can be included in a theory of digitalis action.
The important observations of Lee20 on the action of ouabain on the relaxing factor must be related to mechanisms beyond the membrane and thus may be related to the contracture produced by digitalis. It is known that digitalis can become effective in the virtual absence of extracellular Ca++ but the presence of some Ca++ is essential for the production of digitalis contracture. It is thus probable that digitalis can be bound to the contractile protein or relaxing factor in the absence of extracellular Ca++ but the manifestations of this combination could only occur in the presence of Caf+.
The inhibition of relaxor activity by ouabain observed by Lee20 could be due to: (a) inactivation of the relaxing factor by ouabain, (b) ouabain may convert inactive or unavailable Ca++ to an active or available form, (c) ouabain may prevent the attachment of the soluble relaxing substance to the contractile protein. The data on hand to not allow the expression of preferences for any of these alternatives.
These observations of Lee should initiate some fruitful experimental approaches to the study of digitalis pharmacology. It would be important to see whether this effect of ouabain can be repeated with other active cardiac glycosides and whether it is specific for the cardio-active forms of digitalis. It is hoped that such methodology and other new approaches may give us further insight into the cellular action of digitalis and its interaction with Ca++ and may soon clarify some of the problems alluded to in this paper.
ACKNOWLEDGMENT
We would like to thank Dr. Henrotte of Liege, Belgium for allowings us to read a manuscript entitled “Les ions alcalino-ter-reux et la muscle cardiaque”, which will be published in Heffter-Heubner Handbuch der Experimentellen Pharmakologie.
REFERENCES
1. G. A. Stewart, J. Pharm., Lond. 10, 74 (1958).
2. S. Hajdu and E. Leonard, Pharmacol. Rev. 11, 173 (1959).
3. K. S. Lee, D. H. Yu, D. I. Lee and R. Burstein, J. Pharmacol. 132, 139 (1061).
4. R. Tuttle, P. N. Witt and A. Farah, J. Pharmacol. 133, 281 (1961).
Cardiac glycosides and calcium
169
5. P. K. Boyer and C. A. Poindexter., Amer. Heart J. 20, 580 (1940).
6. N. E. Clarke and R. E. Moscher, Circulation 5, 907 (1952).
7. A. J. Clark, Proc. Roy. Soc. Med. 5, 1 (1915).
8. F. Ransom, J. Physiol. 51, 176 (1917).
9. A. von Konschegg, Arch. Exp. Path. Pharmak. 71, 251 (1913).
10. O. Loewi, Arch. Exp. Path. Pharmak. 82, 131 (1918).
11 . L. Lendle, Heffters Handbuch der Experimentellen Pharmakologie Ergänunzungwerk 1. Springer-Verlag, Berlin, Göttingen, Heidelberg (1935).
12. N. Wershinin, Arch. Exp. Path. Pharm,ak. 63, 386 (1910).
13. H. Gold and D. J. Edwards, Amer. Heart J. 3, 45 (1927).
14. R. A. McGuigan and J. A. Higgins, J. Lab. Clin. Med. 23, 839 (1937).
15. W. Wilbrandt, Wiener Klin. Wschr. 108, 809 (1958).
16. W. T. Salter, L. J. Sciarini and J. Gemmel, J. Pharmacol. 96, 372 (1949).
17. S. Weidmann, J. Physiol. 129, 568 (1955).
18. J. Dudel and W. Trautwein, Arch. Exp. Path. Pharmak. 232, 393 (1958).
19. W. Trautwein and P. N. Witt, Arch. Exp. Path. Pharmak. 216, 197 (1952).
20. K. S. Lee, J. Pharmacol. 132, 149 (1961).
21. R. Tuttle, and A. Farah, J. Pharmacol. 135, 142 (1962).
22. V. Kruta, C. R. Soc. Biol., Paris 124, 791 (1938).
23. D. Teiger, Unpublished observations.
24. V. Weizsäcker, Arch. Exp. Path. Pharmak. 72, 282 (1912).
25. W. Wilbrandt, K. Brawand’ and P. N. Witt, Arch. Exp. Path. Pharmak. 219, 397 (1953).
26. P. N. Sanyal and P. R. Saunders, J. Pharmacol. 122, 499 (1958).
27. H. J. Schatzmann, Helv. Physiol. Acta. 11, 346 (1953).
28. E. H. Wood and G. K. Moe, Amer. J. Physiol. 123, 219 (1938).
29. R. L. Vick and J. B. Kahn, J. Pharmacol. 121, 389 (1957).
30. M. K. Cattell and H. Goodell, Science 86, 106 (1937).
31. H J. Schatzmann and P. N. Witt, J. Pharmacol. 112, 501 (1954).
32. P. Lundsgaard-Hansen, Arch. Exp. Path. Pharmak. 231, 577 (1957).
33. P. N. Witt, J. Pharmacol. 119, 195 (1957).
34. M. Reiter, Pflügers Arch. 267, 158 (1958).
35. F. Sulser, Quoted from W. Wilbrandt, Wiener Med. Wschr. 108, 809 (1958).
36. W. Wilbrandt and H. Koller, Helv. Physiol. Acta 6, 208 (1948).
37. H. C. Lüttgau and R. Niedergerke, J. Physiol. 143, 486 (1958).
38. R. Niedergerke, Experientia 15, 128 (1959).
39. R. Niedergerke and E. J. Harris, Nature, Lond. 179, 1068 (1957).
40. W. Wilbrandt and R. Caviezel, Helv. Physiol. Acta 12, C40 (1954).
41. S. C. Harvey and E. E. Daniel, J. Pharmacol. 106, 394 (1952).
42. L. Thomas Jr., W. Jolley and R. Grechman, Fed. Proc. 17, 162 (1958).
A. Farah and P. N. Witt
170
42a. A. A. Sekul and W. C. Holland, Amer. J. Physiol. 197, 757 (1959)
43. O. Loewi, J. Mt. Sinai Hosp. 19, 1 (1952).
44. O. Loewi, J. Pharmacol. 114, 90 (1955).
45. B. Katzung, H. Rosin and F. Scheider, J. Pharmacol. 120, 324 (1957),
46. A. J. Clark, J. Physiol. 47, 66 (1913).
47. O. Loewi, J. Pharmacol. 96, 295 (1949).
48. R. A. Peters, Proc. Roy. Soc. B 139, 143 (1952).
49. S. Hajdu and E. Leonard, Circ. Res. 6, 740 (1948).
50. W. F. H. M. Mommaerts, B. C. Abbott and W. J. Whalen, Structure and Function of Muscle (edited by G. H. Bourne), vol. 2, p. 517. Academic Press, New York and London (1960).
51. S. Hajdu, Amer. J. Physiol. 174, 371 (1953).
52. R. F. Furchgott and T. de GubarofF, J. Pharmacol. 124, 203 (1958).
53. V. Kruta, Arch. Int. Physiol. 45, 332 (1937).
54. P. Braveny and V. Kruta, Arch. Int. Physiol. 66, 633 (1958).
55. O. Langendorff, Arch. Physiol. Lpz. 284, 287 (1885).
56. H. Rosin and A. Farah, Amer. J. Physiol. 180, 75 (1955).
57. B. Katzung and A. Farah, Amer. J. Physiol. 184, 557 (1956).
58. W. Trautwein, Herzinsuffizienz und Digitalis Wirkung, 114. Springer-Verlag, Berlin, Göttingen, Heidelberg (1959).
59. A. Farah and T. A. Loomis, Circulation 2, 742 (1950).
60. R. Mendez and C. Mendez, J. Pharm,acol. 107, 24 (1953).
61. R. Niedergerke, J. Physiol. 134, 569 (1956).
62. A. A. Siebens, B. F. Hoffman, P. F. Cranefield and C. Me. Brooks, Amer. J. Physiol. 197, 971 (1959).
63. O. Loewi, Arch. Exp. Path. Pharmak. 83, 366 (1917).
64. M. Moulin and W. Wilbrandt, Experientio. 11, 72 (J955).
65. R. Niedergerke, J. Physiol. 138, 506 (1957).
66. J. G. E. Henrotte, E. Cosmos and W. O. Fenn, Amer. J. Physiol. 199, 779 (1960).
67. P. N. Witt, Unpublished observations.
68. C. P. Bianchi and A. M. Shanes, J. Gen. Physiol. 42, 803 (1959).
69. E. Cosmos, Amer. J. Physiol. 195, 705 (1958).
70. A. A. Sekul and W. C. Holland, Amer. J. Physiol. 197, 752 (1959).
71. R. Niedergerke, J. Physiol. 134, 569 (1956).
72. W. Wilbrandt and H. Koller, Helv. Physiol. Acta 6, 208 (1948).
73. M. H. Draper and S. Weidmann, J. Physiol. 115, 74 (1951).
74. A. S. V. Burgen and K. G. Terroux, J. Physiol. 119, 139 (1953).
75. T. C. West and D. W. Amory, J. Pharmacol. 130, 183 (1960).
76. B. F. Hoffman and P. F. Cranefield, Electrophysiology of the Heart, p. 37, McGraw-Hill, New York (1960).
77. W. Trautwein and J. Dudel, Pflügers Arch. 260, 24 (1954).
78. P. N. Sanyal and P. R. Saunders, Proc. See. Exp. Biol., N. Y. 95, 156 (1957).
79. R. Caviezel and W. Wilbrandt, Helv. Physiol. Ada 16, 22 (1958).
Cardiac glycosides and calcium
171
80. J. M. Benforado, J. Pharmacol. 122, 100 (1958).
81. M. Taeschler and H. Weidmann, Helv. Physiol. Acta 15, C81 (1957).
82. B. Frankenhauser, J. Physiol. 137, 245 (1957).
83. C. R. Mines, J. Physiol. 46, 188 (1913).
84. W. Trautwein and K. Zink, Pflügers Arch. 256, 68 (1952).
85. B. B. Marsh, Nature, Lond. 167, 1065 (1951).
86. J. R. Bend all, Nature, Lond. 170, 1058 (1952).
87. E. Bozler, J. Cen. Physiol. 38, 149 (1954).
88. E. Bozler, J. Gen. Physiol. 38, 735 (1955).
89. C. J. Parker and J. Gergely, J. Biol. Chem. 236, 411 (1961).